
Highly multiplexed reference materials are particularly valuable when developing and optimizing new NGS assays because they allow you to evaluate the performance of your assay across a large number of variants including different variant types (SNVs, indels, homopolymeric variants, etc.) and contexts. However, it can be frustrating when a variant in the reference material is not detected, or not detected at the expected variant allele frequency. Troubleshooting such issues can give new insight into the performance of the assay. Here we share some stories from Seraseq™ users where the lack of detection of one or more variants at the expected levels helped them improve their assay or set more appropriate QC thresholds.
Check for Assay and Variant Compatibility
Confusion about what variants different NGS targeted panels evaluate is actually a common reason for non-detection. While it might seem obvious what genes and variants a particular assay targets, in fact, it can often be challenging to find this information. For example, SeraCare’s Seraseq™ Myeloid Mutation DNA Mix contains two JAK2 variants: the c.1849G>T mutation encoding the p.V617F variant as well as the c.1624_1629delAATGAA mutation encoding the p.N542_E543del variant. A customer was validating the ArcherDX Core AML kit and able to detect the p.V617F variant, but not the p.N542_E543del variant. This mutation is located at chr9:5070035-5070040, a region of JAK2 which is not evaluated in the Core AML kit. Finding this information may require a deep dive into the assay product literature—in this case, the Target Region GTF file provided by ArcherDx for this assay. JAK2 exon 12 mutations, such as c.1624_1629delAATGAA, result in a myeloproliferative phenotype important in patients diagnosed with polycythemia vera or idiopathic erythrocytosis (The New England journal of medicine 2007;356(5):459-68 ISSN:1533-4406, PUBMED:17267906). The insight gained from the reference material may prompt the customer’s lab to look into expansion or customization of the NGS panel, if detection of these variants is important to meet their testing requirements.
Sequence Reference Materials at Sufficient Depth
Non-detection, or poor detection of variants can also result when there is low sequencing depth. In order to monitor the accuracy and precision of test performance, the variance of the reference material should be minimized so that other sources of variance that affect the test can be identified. This means that a reference material must be tested with sufficient input at a sufficient coverage depth. It is not recommended to perform low pass sequencing of the reference material in order to save sequencing space for the unknowns. Figure 1 shows modeling of the expected results for Seraseq Tumor Mutation Mix AF10 at various read depths. For example, 2000 reads of a variant present at 10% in the library will result in an observed frequency of between 8.7% and 11.4%, 95% of the time. Decreasing the coverage depth to only 500 reads substantially broadens the 95% confidence interval.
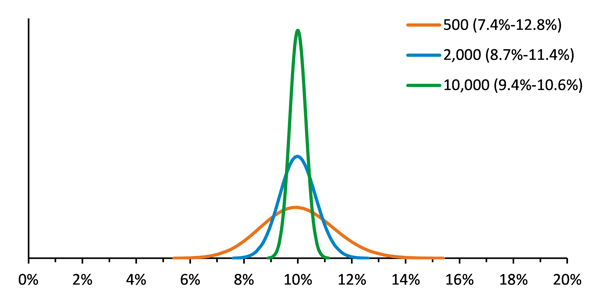
This modeling helped us to understand why a customer using SeraCare’s Seraseq™ Myeloid Mutation DNA Mix was observing the NPM1 p.W288fs*12 insertion mutation at only ~3%, when the specified allele frequency is 5%. She was using an ArcherDx AML VariantPlex assay and, although she was using input DNA amounts in the recommended range (~50 ng), the overall depth was quite low, which can lead to poor precision. The NPM1 p.W288fs*12 insertion variant was present in the raw data with 440 REF and 14 ALT observations. Further investigation showed there were only 239,129 fragments (and only 21,589 unique fragments), which is below the assay manufacturer’s suggested read depth. Having only 10% of fragments be unique suggests that library synthesis was inefficient. The enzymatic fragmentation used in VariantPlex is sensitive to time and input amount and these often require optimization. Inadvertent overdigestion may result in fragments that are too short to be useful, and these short fragments may be removed during subsequent AMPure bead cleanup steps, resulting in a library with low overall complexity. This insight gained from the reference material prompted the customer to review procedures for enzymatic fragmentation and to troubleshoot ways to increase library conversion efficiency and sequencing depth.
Generate Sequencing Libraries with Sufficient Complexity
Similar problems can occur when insufficient amounts of input DNA or RNA are used for library preparation. A customer was having trouble detecting the FGFR3 S249C variant in Seraseq ctDNA Mutation Mix v2 at the expected allele frequency: the variant was detected at a very low level of 0.3% in the AF2% material. Investigation showed this may be an issue of sampling with small numbers. The customer was using an input amount of 10 ng and an amplicon-based assay. Based on the observed amplicon lengths, the expected number of amplifiable molecules with this input amount was at or below 500. At that point, failure to observe very low variant frequency percentages is possible by chance since a 2% sample would average about 10 mutant molecules and a 1% sample only 5 mutant molecules. Getting 5 or 0 (or 20) is also possible due to the stochastic nature of sampling small numbers. In this case, using a higher input quantity of DNA is likely to result in greater precision around the expected values.
Amplicon Size can Limit Sequencing Library Complexity
Finally, in amplicon-based NGS assays, the size of the amplicon generated for detection of a particular variant can make a difference when assessing variants in ctDNA or other fragmented formats. A customer was using SeraCare’s Seraseq™ ctDNA Reference Material AF2% and extracting 2 mL of the full process reference material (of the 5 mL provided). He was able to detect most of the EGFR variants in the material (ex19del, L858R, ex20ins), but could not detect T790M mutations and the question was why is there detection of some, but not other variants? “Back-of-the-envelope” calculations gave some clues. Typically, there is 1 germline copy of a particular single copy gene sequence per ~3.5 picograms of DNA. For a mutant sequence present at 2% variant allele frequency mutations, there is 1 copy per ~175 picograms of DNA (50x less). With ~50 ng of ctDNA yield typically extracted from 2 ml, there would be ~286 mutant copies. Given the fragmented nature of cfDNA, not all of those mutant copies will be amplifiable since both primer binding sites have to be present; so there should be <286 amplifiable mutant copies per 2 ml. In fact, a typical size for amplicons in some amplicon-based assays that are being applied to ctDNA is 120 – 160 bp. The Seraseq ctDNA products have fragment sizes of approximately 165 bp typical of native cell free DNA in plasma, so with fragment sizes and 120-160 bp amplicons, only 3% to 27% of input will be amplifiable. Thus 50 ng would perform no better than 1.5 to 13.5 ng of genomic DNA input, which would result in low precision. Furthermore, slight differences in the size of the amplicons to detect different mutations might further skew results towards greater detection of some variants (with smaller amplicon sizes) and less detection of other variants (with larger amplicon sizes) possibly explaining the customers failure to detect EGFR T790M.
Generating Insight
In this FAQ, we’ve seen examples of variants in reference materials that were not detected at their expected levels and looked into the issues that can generate this variability. The recommendations when evaluating a reference material:
- Use a compatible assay. We recommend reviewing the variant details in product literature from the assay manufacturer as well as the variant details of the Seraseq material to ensure compatibility.
- Sequence at a high coverage depth. Most assay manufacturers will recommend a particular read depth for their assay. It is best to follow those recommendations to ensure better confidence in the data.
- Use sufficient amplifiable DNA to generate a library with high complexity. When working with low input DNA amounts and/or low AFs of variants, it is important to consider how much of the DNA can be amplified.
However, when testing patient samples, some parameters, such as input DNA amounts, may be fixed and the reference materials should be tested in the same way as the unknowns. The performance of the reference material under these conditions gives insight into the strengths and weaknesses of the assay. The variability of the reference material informs on the variability of the assay due to small number sampling in low complexity libraries or low coverage depth. The highly multiplexed format of Seraseq reference materials gives insight into variant to variant differences in detection and may aid in assay optimization.
Are you looking for clarity on validation guidance?

Our new eBook offers insights for more effective and less costly assay validation and will walk you through key considerations and guidelines for success.
"Nothing is more frustrating than finding a sample that is positive for a relevant variant but cannot be tested multiple times due to sample depletion."

