
 As originally seen in The Journal of Precision Medicine March 2019.
As originally seen in The Journal of Precision Medicine March 2019.
Rapid innovation has had a dramatic impact on reducing the cost of DNA sequencing. This has enabled laboratories of all sizes across the developed world to have access to one of the most powerful diagnostic modalities in human history to aid in the treatment of some of our most challenging health conditions.
However, while we can all acknowledge that technological innovation with NGS has pushed the envelope towards producing more answers, operational excellence is now needed to ensure that we are producing the right answers, consistently and reproducibly. This means that a given NGS assay must produce the same answers from technologist to technologist, instrument to instrument, within the same laboratories, and across different laboratories. There are three primary reasons why operational excellence is still challenging for laboratories utilizing clinical NGS: the workflow complexity of NGS; a shortage of IVD assays and components; and a scarcity of support. These factors have significant impacts on the ability for laboratories to achieve consistent, reproducible results, and are described in detail in this article.
The first reason why reproducibility is challenging for clinical NGS is the high degree of complexity of the NGS workflow (see workflow depicted in Figure 1). Nucleic acid (NA) extraction, NA library preparation, sequencing, primary analysis, secondary analysis, clinical interpretation, and reporting can take up to a week to complete. The wet bench component alone can take over 10 hours of hands-on time, and often includes several ancillary pieces of equipment each having their own research use only (RUO) reagents and consumables.
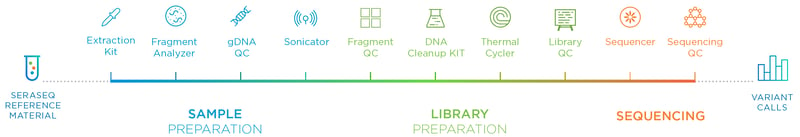 Figure 1: Overview of the NGS Workflow. The workflow can take up to 3 days to complete and involves many steps.
Figure 1: Overview of the NGS Workflow. The workflow can take up to 3 days to complete and involves many steps.
Technician experience, instrument performance, PCR duplicates, cluster density, chip loading, and numerous other factors can all conspire individually or together to alter the performance of the assay. At the same time, the quality of samples a laboratory receives can be highly variable and contribute to erroneous results due to the process of preserving samples for subsequent analysis or tumor heterogeneity.
The second reason is that while it is encouraging that the FDA is accelerating their efforts to clear NGS-based IVDs as companion diagnostics, most of the laboratories (numbering in the thousands) are conducting clinical NGS testing using RUO platforms and reagents in the form of laboratory-developed tests (LDTs). These RUO platforms and reagents were not developed as fit-for-purpose clinical solutions for a specific intended use. Many of these laboratories lack the framework (e.g., validated quality systems, procedures, policies, and documentation) that the FDA requires for companies to market and support their products as IVDs. As a result, clinical genomics laboratories seeking to apply the power of clinical NGS are required to gap-fill these requirements by developing their own quality management programs.
These RUO platforms, reagents, and inhouse framework (or lack thereof) can pose challenges in a clinical testing environment with respect to tracking key performance metrics, such as sequencing quality (Q Score). A sequencing run of “75% of bases greater than Q30” means that 75% of the base calls have an accuracy of at least 99.9%. The higher the percentage, the better the sequencing quality. The lower the percentage, the lower the quality, which can impact the final base call results.
Figure 2 is a hypothetical example of how a clinical genomics laboratory conducts validation studies spanning several years over the lifecycle of an RUO sequencing consumable. Initially, “User X” used three lots to validate their assay. Later, “User Y” conducted a second validation. Since the “Bases Called % > Q30” was higher, there was no issue. Later, even though the sequencing consumable still met its RUO specifications, “User Y” failed their validation. For this scenario not to have occurred, the reagent must have undergone appropriate studies by the manufacturer to establish the long-term performance variability across multiple lots. Studies such as this are often required as part of an IVD submission but may not be in place for an RUO product.
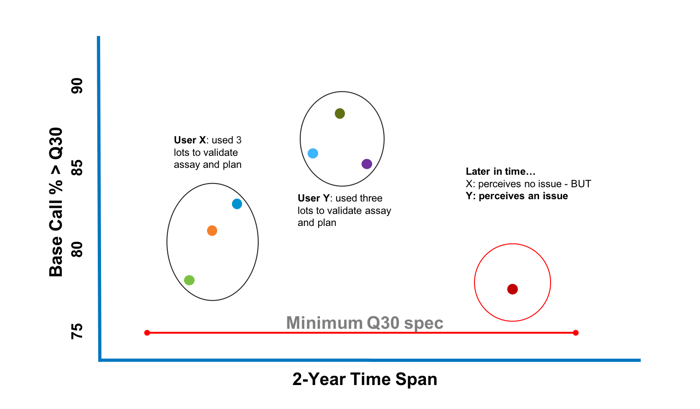
Figure 2: Schematic of how RUO reagent performance can drift over time negatively impacting clinical laboratory validation
even though performance meets manufacturing specifications for an RUO designated product.
The third reason is the lack of direct support from manufacturers for customers to validate and operationalize in part due to the RUO labeling the products are sold under. As a result, laboratories wishing to utilize clinical NGS must sort out all of the underlying processes and procedures on their own, without alignment on best practices, resulting in a lack of standardization across clinical NGS laboratories.
Because of these challenges and the shift of the operational burden to each individual laboratory, it is of vital importance that laboratories utilizing clinical NGS develop suitable quality control (QC) strategies and programs. Otherwise, unintended variability of testing results can put patients at risk for false positives or false negatives resulting in patients not getting the appropriate treatments when they should – or getting treated when they shouldn’t.
According to the survey, 29% of laboratories performing clinical NGS use positive controls only on new lots of reagents, or not at all, while 71% of laboratories report that they use positive controls every few runs (see Chart 1).
Laboratories typically establish the sensitivity, specificity, and limit of detection of an NGS assay during analytical and clinical validation. However, the survey reveals that many laboratories are not monitoring assay performance in real time for each sequencing run as part of a robust quality control program. In other words, laboratories are not ensuring that the ongoing performance of their assay Figure 1: Overview of the NGS Workflow. The workflow can take up to 3 days to complete and involves many steps. is on par with what was established after their validation efforts were completed. One underlying reason for this is cost, but the other is that accreditation guidelines such as CLIA and CAP do not specify the frequency or the type of run controls that are required for a robust QC program. Instead, these laboratories rely on laboratory directors to determine what is appropriate for the assay based on the data gathered during validation.
Many clinical NGS laboratories are using remnant clinical samples found in their respective institutions, or mixed cell lines as substitutes for reference materials. There are many challenges associated with this practice. First, a QC management program needs a consistent source of reference materials that can be used to monitor instrument performance longitudinally. Remnant samples are often quickly exhausted, requiring the laboratory to find other remnant clinical samples to continue their QC efforts which inhibits the ability to establish a reliable longitudinal baseline. The quality of remnant samples may also change over time as the sample becomes depleted and goes through multiple freeze/thaw cycles. Second, both remnant samples and cell lines do not contain the different types of variants (e.g. SNVs, insertions, deletions, copy numbers, fusions) that represent the biology a clinical NGS assay is seeking to interrogate. For example, a 100 gene panel used in NGS requires more than a handful of variants to be monitored as part of a robust QC program in order to offer confidence in the fidelity of results. Finally, homebrew reference materials are not manufactured under a robust quality management system to guard against variability over time and cannot provide a true sustainable “gold standard” to reliably measure assay performance against (see Chart 2).
Other laboratories use sequencing metrics as surrogates for positive controls.
The survey reveals that many laboratories use sequencing metrics generated from clinical samples as a surrogate for internal positive controls. The top 5 metrics cited as used for this purpose were read depth, DNA library quantification, quality score, DNA concentration, and percent (%) reads passing.
Figure 3 shows the risk with this approach. In this study, 180 runs were performed and 10% of those runs had false positives or false negatives detected in the internal run control. 40% of the detected false positives and false negatives did not have any failed statistical metrics that would have promoted additional investigation. Had an internal run control not been used, there would have been a risk for this laboratory to have potentially reported out false positives and negatives.
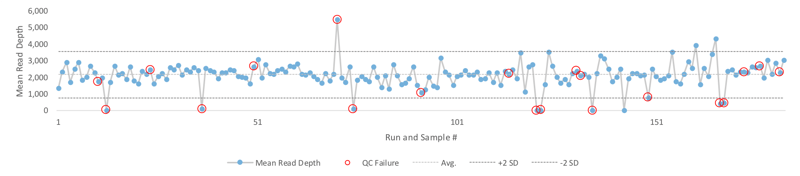 Figure 3: A study comprising of 180 NGS runs resulted in 10% false positives or false negatives (red circles). 40% of those runs with false positives or false negatives did not have a failed mean read depth (defined as greater than 2 SD) and were only detected using internal positive controls (red circles within 2 SD).
Figure 3: A study comprising of 180 NGS runs resulted in 10% false positives or false negatives (red circles). 40% of those runs with false positives or false negatives did not have a failed mean read depth (defined as greater than 2 SD) and were only detected using internal positive controls (red circles within 2 SD).
From this, we can conclude that the use of statistical metrics as a substitute for in run controls are not enough to guard against false positives and false negatives. Pharma stakeholders would be well advised to audit the quality management programs that clinical NGS laboratories are using specifically in terms of how and when a laboratory uses positive controls; as well as whether the laboratory is using commercially available reference materials manufactured with a robust quality management system as part of their QC management strategy.
Maximizing therapeutic benefit through global operational excellence requires the right tools – NTRK Fusion Case Study
Bayer has recently completed initial development of Vitrakvi® (larotrectinib) that targets patients with TRK (Tyrosine Kinase Receptor) fusion cancers. TRK Fusion proteins are a primary oncogenic driver across multiple tumors in adults and children. In TRK fusion cancer, the NTRK gene fuses with an unrelated gene causing overexpression of the TRK protein and uncontrolled cell growth. Because many different fusions can cause this overexpression, NGS is an ideal platform to look for these important tumor biomarkers by looking at many possible gene fusion pairings.
A key challenge for laboratories looking to detect NTRK (neurotrophic tyrosine receptor kinase) gene fusions with their NGS tests is the scarcity of characterized clinical samples for the various fusion genes, making the use of remnant samples extremely challenging. SeraCare, together with scientists at Bayer, have developed a representative panel of NTRK RNA fusion reference materials to enable laboratories to reliably detect some of these important fusion genes across NTRK 1, 2, and 3.
A key benefit associated with the development of reference materials like this is that it enables proficiency testing organizations to ensure that laboratories are achieving consistent results – from instrument to instrument, operator to operator, and laboratory to laboratory. This enables more laboratories to conduct reliable, accurate testing, which in turn, broadens the reach of therapeutic intervention to patients who might not ordinarily have access. Without a single, unifying reference material, this would be almost impossible.
In short, commercially available reference materials are a key enabler of operational excellence which enables pharma to increase their reach by accessing patient populations which would otherwise be missed.
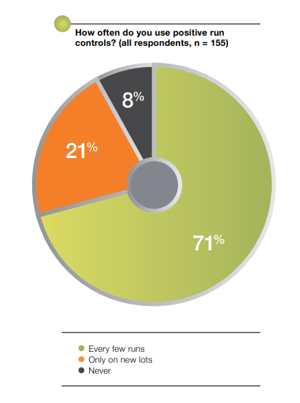
Chart 1: A first of its kind survey1 was recently conducted to better understand how clinical NGS laboratories address quality control, and the results demonstrate the symptoms of the challenges outlined above and provide a picture of what areas require further development by the field.
Most laboratories utilizing clinical NGS under CLIA guidelines do not use positive controls in every run.
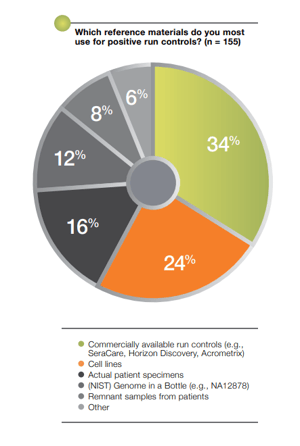
Chart 2: Out of the laboratories that do use positive controls, 66% of them use “homebrew” approaches.
Final thoughts
We are continuing to make rapid technological advances in our efforts to understand and treat our most challenging health conditions. However, technological innovation is just one side of the coin with respect to the application of clinical NGS diagnostics for personalized medicine. Genomic platform providers, the FDA, accreditation bodies, pharma stakeholders, laboratory directors, and managed care providers must all hold each other accountable regarding operational excellence in order to realize the true promise of personalized medicine.
Sources
- Trends in Clinical NGS QC Management A Practitioner Survey. September, 2018.
Trends in Clinical NGS QC Management:
Expert Insights to Ensure Quality Results for Your Lab
Recorded Webinar and Survey Report
Learn how QC has been implemented by your peers as the panel expands on the results of
the most comprehensive survey to date on NGS QC practices.

