
DNA methylation is an epigenetic mechanism modulating the function and expression of genes. DNA methylation regulates gene expression by recruiting proteins involved in gene repression or by inhibiting the binding of transcription factor(s) to DNA. Changes in DNA methylation are associated with numerous diseases and abnormal DNA methylation has been implicated as one of the mechanisms driving tumor onset, development, progression and recurrence. Identification of tumor-associated methylation patterns is of great diagnostic potential, especially for early detection of disease.
In Part 2 of this two-part blog series, the article continues to review the methylated DNA biomarkers in plasma webinar. As a reminder, Part 1 covered the main themes discussed by our invited webinar speakers - click here to read that article.
To conclude the recent webinar hosted by LGC Clinical Diagnostics and GenomeWeb, Dr. Yves Konigshofer talked about levels of evidence required for clinical diagnostics, from analytical validity through clinical validity and clinical utility. Reference materials can help prove analytical validity and assess the impact of pre-analytical factors. Methylation reference materials that he is currently developing at LGC CDx address the analytical validity of 5’methylcytosine measurements in cfDNA.
Traditional methylation analysis is challenging, because bisulphite conversion can skew the data depending on the degree of conversion, so optimization is needed to balance false negatives and positives. Dr. Konigshofer and his team are working on developing a set of reference materials with a range of 0% - 100% methylation, using dPCR at methylated SNPs to assess the level of methylation at individual sites before confirming global methylation levels with whole genome sequencing. When comparing WGS with bisulphite sequencing data, he noticed the latter show regional coverage bias. Does this mean that bisulphite is damaging certain regions more? Or does the choice of dU tolerant polymerase have an effect?
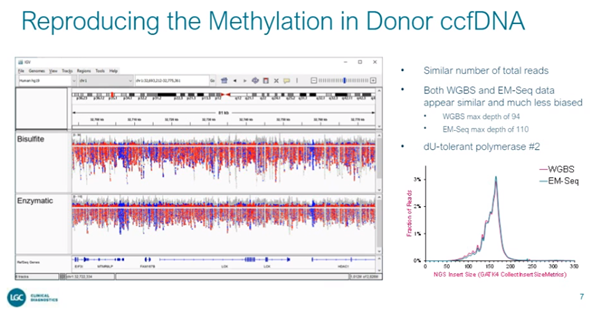
External testing of prototype reference materials confirmed expected levels of methylation and the viability of preparing a precise ladder by serial blending, with one lab observing discrepancies at 50% methylation, highlighting potential differences in the assay used. They are now moving to the next step, which is reproducing the methylation patterns of donor-derived ccfDNA. The initial data is promising, showing that using another dU tolerant polymerase indeed helps maintain coverage uniformity.
Questions & Answers
The webinar garnered an unprecedented number of questions from the audience, ranging from the technical to the clinical. While most of them were addressed live, Dr. Dennis Lo (DL) and Dr. Yves Konighshofer (YK) kindly answered additional questions posted by the attendees relevant to his work.
Is there any benefit in super low ng-pg sensivity for methylation studies? Is there a limit of value? Avida got down to 10 ng but not sure if lower is always better.
DL: I think that this will depend on the specificity of the methylation marker that is being detected. Methylation markers are rarely as ‘black and white’ as genetic markers like single nucleotide changes.
YK: When the goal is to detect altered methylation and one is reasonably sure that a particular genomic region is normally completely unmethylated or methylated, then a few molecules that show the opposite may be indicative of something. Since tumor content in ccfDNA is often very low, looking at multiple such regions also increases the likelihood of detecting a change.
Do you expect methylation differences between short ccfDNA (50-200bp) and large ccfDNA? Most ccfDNA is relatively short, so that is the primary focus for methylation differences, correct?
DL: The advantage of large ccfDNA is that one may be able to determine a ‘methylation haplotype’ even from a single molecule. As the field of large ccfDNA is so new, more work is needed to see if there may be methylation differences between short ccfDNA and large ccfDNA.
Tumor samples have been reported to have a larger proportion of fragments below 150 bp (https://www.science.org/doi/10.1126/scitranslmed.aat4921). Is SMRT showing a reverse trend that cfDNA from tumor cells tends to have larger fragments?
DL: Even though long ccfDNA fragments are detectable in cancer patients, but the overall size distribution is still shorter than the size distribution in subjects without cancer.
What is the fraction of high molecular DNA fraction e.g. >1Kb?
DL: The median percentage of cfDNA molecules above 1 kb, as measured using SMRT, is approximately 15%.
Is it necessary to use multiple aligners and methylation callers to account for biases before drawing conclusions?
DL: Currently we use the BWA aligner for our long ccfDNA work. For methylation calling, we only use our “holistic kinetic model”.
YK: It can be helpful. With bisulphite conversion, every single CG in the genome could end up being sequenced as CG or TG, which adds a lot of potential diversity around possible DNA sequences, and different aligners handle this differently.
What is the best reference material to use to mock methylated cfDNA?
DL: we use M.Sss1 treated DNA as our methylated control and whole genome amplified DNA as our non-methylated control.
YK: The methylated ctDNA ladder currently under development at LGC CDx should provide the best solution, incorporating both methylated an unmethylated control.
It would be interesting to simultaneously study nucleosome positioning and methylation profile of cfDNA fragments to increase the performance of the test and learn new biological insights. Could you comment?
YK: The impact of nucleosome positioning on ccfDNA fragments has been referred to as the fragmentome. We try to reproduce this in some of our amplified donor-derived reference materials. Right now, we are focused primarily on supporting the determination of analytical validity around the measurement of methylation in ccfDNA.
In ctDNA-derived reads, are there more reads from repeat regions or unique regions? Where do you see more methylation differences?
DL: One can see methylation differences between cfDNA from cancer patients and non-cancer subjects from both repeat regions (e.g. Alu) and certain unique regions.
How stable is the methylation status of cfDNA and what influence does the small size of cfDNA have on methylation analysis?
DL: I believe that cfDNA reflects the disease status of a cancer patient. Hence, the methylation profile of cfDNA would change with progression, or response to treatment. The small size of cfDNA means that for most molecules, the number of CpG sites may be one or even zero.
YK: A small fragment (e.g., as would be found with ccfDNA) can be harder to align unambiguously than a longer one if the sequence therein could map to multiple locations. Additionally, smaller fragments can be harder to recover during NGS library prep where a goal is to remove adapter dimers.
Is single-cell methylation going to be important in near future to address signal to noise that Jimmy mentioned?
DL: I think that single-cell methylation analysis will tell us important biological information regarding the spectrum of methylation changes that we can see in tissues in health and disease.
Does cfDNA isolation using Qiagen isolation of cfDNA retain the 1-3 kb fragments you are talking of?
DL: Yes, the Qiagen methodology does retain those molecules.
How significant are the fragment end motifs in cancer detection?
DL: I believe that end motifs are a valuable class of cfDNA fragmentomic markers.
Watch Now
Watch the webinar now by clicking here, or on the button below.

