
Clinical labs must constantly evolve their test offerings in order to support the most recent advances in clinical care. For next-generation sequencing (NGS) tumor profiling assays, there are often multiple commercially available kits with similar claims for gene content and sensitivity, as well as customized solutions. How can you quickly perform an effective evaluation of available assay systems to make a data-driven choice?
Many factors contribute to the decision of which off-the-shelf "research use only" assays or customized solutions will best meet the needs of the lab, clinicians, and patient population. Kit costs, capital equipment expenditures, complexity of workflow, and turnaround time are all important factors that can be relatively easily compared and assessed between assay systems. However, the key parameters for determining effective, personalized treatment for patients are accuracy, reproducibility, and sensitivity of the assay, and these can be much more challenging to critically evaluate but must be rigorously validated1-4. However, use of a highly multiplexed, consistent, and well characterized reference material can greatly facilitate the comparison of assay systems5.
Assessing Accuracy
Not all variants perform the same in each assay because of factors such as different primer designs, different variant callers, and different sequencing density over the region of interest6. Determining accuracy, the ability of the assay to detect all the variants it claims to detect, is therefore of critical importance. While assessing detection of every mutation contained in the panel’s gene content isn’t practical, evaluation of those mutations most critical to the patient population is important. How do you verify that all clinically important variants are detected appropriately when using remnant patient samples or cell lines with only one or a few mutations present? Very highly multiplexed reference materials offer the ability to look across many different oncogenes and tumor suppressor genes at a wide variety of difficult variant types, such as medium to large size indels and indels in homopolymeric regions.
For example, the Seraseq Tumor Mutation Mix v2 AF10 was evaluated on market leading cancer hotspot panels run on both the Illumina MiSeq and Ion Torrent PGM instruments. For the vast majority of SNVs and indels, detection and allele frequency across platforms was similar. However, TP53 c. 263delC was not detected on one of the platforms. This mutation, important in cancers in the upper aerodigestive tract, as well as head and neck cancers, is a single nucleotide deletion, but is a deletion of a C in a homopolymer of C’s in a very GC rich region. Further investigation showed that the non-detection was due to prediction modeling used for variant calling: the software may identify candidate variants as likely false positive variants due to systematic read errors resulting from the sequencing polymerase interacting with homopolymer, and therefore not report them. However, for TP53 c. 263delC, this filtering flags a true variant as a false positive, and therefore results in an inability of this system under these parameters, to detect this variant in patient samples. While this problem may be overcome by adjusting filtering thresholds and optimization, the need for this additional development work may impact the assay choice.
Evaluating Precision and Robustness
Knowing the day-to-day, operator-to-operator, and reagent kit lot-to-kit lot variability of an assay can also play into the assay selection choice. Understanding the assay variability requires a consistent sample to be evaluated on multiple runs. Unfortunately for ctDNA NGS sequencing panels, obtaining enough extracted cell-free DNA material from residual patient material is not usually possible, and sheared cell line material is not sufficiently similar to patient samples for this purpose. Use of a whole process reference material that is highly similar to extracted patient cfDNA can be highly useful in evaluating assay variability.
For example, the Archer® Reveal ctDNA™ is an RUO kit widely validated for clinical use. The kit protocol states that successful libraries have been produced with between 1 ng and 300 ng of total DNA mass7. However, how much DNA must be used for reliable variant calling in a clinical setting, and what is the minimum detectable allele frequency at the maximal input DNA mass? What about the minimal input DNA mass? Seraseq ctDNA Mutation Mix v2 (AF1%) and (AF0.25%) were tested to show how highly characterized, consistent reference materials can help answer these questions. The mutations in the reference material are characterized by digital PCR (dPCR) analysis, an absolute quantitation method that precisely defines the allele frequency for each variant. This dPCR data is plotted on the X-axis for 20 variants with the corresponding NGS plotted on the vertical Y-axis. When 100 ng of input DNA was used for testing of the 0.25% allele frequency material, all of the variants were detected at allele frequencies very close to the expected 0.25%. However, when only 10 ng of DNA of the 1% AF was used as the input, at least one variant is not detected and there is much more “noise” or imprecision in the data. Often, labs don’t have enough of a consistent reference material to evaluate the loss in robustness as input amounts go down, but with a well characterized reference material, they can determine the variability at different input cut-offs.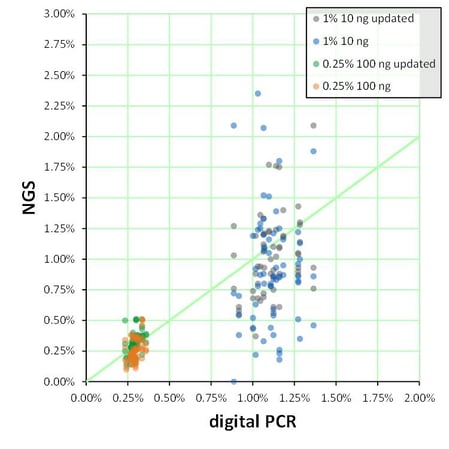
Figure 1: NGS data (vertical axis) was compared to digital PCR data (horizontal axis) for ~20 variants. NGS Testing was performed on two independent runs using the Archer Reveal ctDNA™ Assay and two slightly different workflows (original and “updated”).
Determining Sensitivity
NGS assays are generally qualitative and NGS assays for oncogenic fusion RNAs report either detection or non-detection of fusion targets. Kit manufacturers may provide information about the number of unique NGS reads required for a positive call, but it may be very difficult to correlate this with the number of variant molecules in the patient sample. Such a correlation is critical for setting lower limits of detection for clinical assay validation. Here is where highly characterized reference materials can help. Digital PCR is an absolute quantitation method that can provide the ground truth about the concentration or allele frequency in a reference material. This truth set can then be used to evaluate comparative sensitivities of different assays.
A pathologist was looking to establish sensitivity of her assay for a regulatory submission. She used a custom made Seraseq tumor RNA fusion linearity panel. The concentration of total RNA (from GM24385 reference human cell line) in each panel member was consistent, but the level of the sixteen fusion RNA transcripts went down in 5-fold increments from one panel member to the next. During manufacturing, the copies of each fusion RNA fusion target in the Seraseq panel had been verified by reverse transcription digital PCR. Figure 2 illustrates the dPCR verified values for each panel member for the two of the fusion targets as well as the NGS supporting reads. Although the level of CD74-ROS1 and EML4-ALK fusion RNA are at similar levels in the sample, the NGS reads are ~10-fold different and the NGS assay has different lower limits of detection for these two fusion targets. By using the reference material and evaluating the digital PCR quantitation vs. detection on her NGS assay, she was able to estimate the sensitivity of the assay for each fusion RNA she was validating.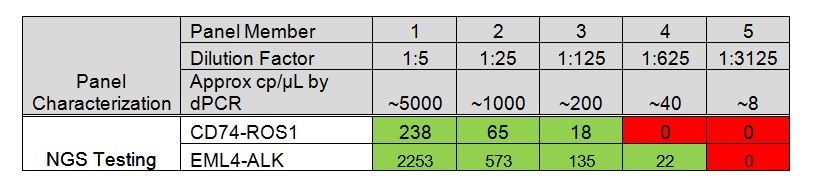
Figure 2: Fusion RNA copies/ µL as determined by digital PCR versus detection on an NGS assay for two fusion targets in the panel. The fusion copies/µl were verified by digital PCR and are shown in grey shading. Green shading indicates that the fusion target was detected and the numbers indicate the NGS supporting reads.
Red shading indicates non-detection.
In summary, a lab cannot assume any RUO-labeled assay detects all the variants at the levels indicated. It is the responsibility of the lab to validate each variant. Highly multiplexed, consistently manufactured reference materials allow you to design an effective evaluation study and make a data-driven decision for new assay development and adoption.
To learn more about these concepts, check out our recent white paper .
References
- Aziz, N., Zhao, Q., Bry, L., Driscoll, D. K., Funke, B., Gibson, J. S., Grody, W. W., Hegde, M. R., Hoeltge, G. A., Leonard, D. G., Merker, J. D., Nagarajan, R., Palicki, L. A., Robetorye, R. S., Schrijver, I., Weck, K. E. & Voelkerding, K. V. (2015). College of American Pathologists' laboratory standards for next-generation sequencing clinical tests. Arch Pathol Lab Med 139, 481-93.
- Gargis, A. S., Kalman, L., Berry, M. W., Bick, D. P., Dimmock, D. P., Hambuch, T., Lu, F., Lyon, E., Voelkerding, K. V., Zehnbauer, B. A., Agarwala, R., Bennett, S. F., Chen, B., Chin, E. L., Compton, J. G., Das, S., Farkas, D. H., Ferber, M. J., Funke, B. H., Furtado, M. R., Ganova-Raeva, L. M., Geigenmuller, U., Gunselman, S. J., Hegde, M. R., Johnson, P. L., Kasarskis, A., Kulkarni, S., Lenk, T., Liu, C. S., Manion, M., Manolio, T. A., Mardis, E. R., Merker, J. D., Rajeevan, M. S., Reese, M. G., Rehm, H. L., Simen, B. B., Yeakley, J. M., Zook, J. M. & Lubin, I. M. (2012). Assuring the quality of next-generation sequencing in clinical laboratory practice. Nat Biotechnol 30, 1033-6.
- Schrijver, I., Aziz, N., Farkas, D. H., Furtado, M., Gonzalez, A. F., Greiner, T. C., Grody, W. W., Hambuch, T., Kalman, L., Kant, J. A., Klein, R. D., Leonard, D. G., Lubin, I. M., Mao, R., Nagan, N., Pratt, V. M., Sobel, M. E., Voelkerding, K. V. & Gibson, J. S. (2012). Opportunities and challenges associated with clinical diagnostic genome sequencing: a report of the Association for Molecular Pathology. J Mol Diagn 14, 525-40.
- Jennings LJ, Arcila ME, Corless C, Kamel-Reid S, Lubin IM, Pfeifer J, Temple-Smolkin RL, Voelkerding KV, and Nikiforova MN. ( 2017).. Guidelines for Validation of Next-Generation-Sequencing-Based Oncology Panels. Journal of Molecular Diagnostics 19 (3), 341–365
- Sims, David J. et al. “Plasmid-Based Materials as Multiplex Quality Controls and Calibrators for Clinical Next-Generation Sequencing Assays.” The Journal of Molecular Diagnostics : JMD3 (2016): 336–349. PMC. Web. 10 Oct. 2017.
- Sarah Sandmann, Aniek O. de Graaf, Mohsen Karimi, Bert A. van der Reijden, Eva Hellström-Lindberg, Joop H. Jansen & Martin Dugas ()Evaluating Variant Calling Tools for Non-Matched Next-Generation Sequencing Data. Scientific Reports 7, Article number: 43169 (2017)
- Archer® Reveal ctDNA Protocol for Illumina, Document number LA090.A Released September 20, 2017. Available at ArcherDx.com
